Нарушения липидного обмена (дислипидемии) влияют на процессы всасывания, трансформации и обмена жиров в организме. Помимо энергетической функции, жиры являются важным компонентом клеточных мембран, участвуют в синтезе гормонов, передаче нервных импульсов и выполняют массу других жизненно важных задач. Поэтому нарушение липидного обмена значительно сказывается на состоянии всего организма в целом и может приводить к развитию тяжелых последствий.
Причины дислипидемии
- Алиментарные – употребление в пищу большого количества животных и растительных жиров. Норма устанавливается индивидуально для каждого человека, средние показатели – от 0,8 до 1 гр. жиров на килограмм массы тела в сутки.
- Врожденные нарушения липидного обмена. Возникают в результате мутаций в генах, которые отвечают за процессы синтеза, расщепления, транспортировки жиров. Эти мутации передаются из поколения в поколение, поэтому заболевание может возникать у маленьких детей без видимых на то причин. Примерами генетических нарушений липидного обмена являются болезнь Гоше, Тея — Сакса, Ниманна — Пика.
- Вторичные нарушения липидного обмена. Обусловлены другими заболеваниями, которые могут затрагивать желудочно-кишечный тракт, эндокринную и ферментную системы, различные внутренние органы (печень, почки).
Среди провоцирующих факторов, которые повышают риск развития нарушений липидного обмена, отмечаются вредные привычки (курение, злоупотребление алкоголем), малоподвижный образ жизни, избыточный вес, хронические стрессы, прием гормональных препаратов.
Как проявляется нарушение липидного обмена
На первый взгляд может показаться, что нарушение липидного обмена обязательно проявляется в виде избыточного отложения подкожного жира на различных участках тела. Данный симптом может развиваться, но он не является специфичным, а характерен для многих других состояний. Клиническая картина дислипидемии зависит от того, повышена или понижена концентрация жиров в крови. В первом случае отмечаются следующие признаки:
- появление отложений холестерина на веках, животе, лице, конечностях. Внешне они выглядят как желтоватые пятна, несколько возвышающиеся над поверхностью кожи;
- атеросклероз сосудов, который сопровождается болью в сердце, нарушением дыхания, изменением сердечного ритма и другими признаками;
- увеличение печени и селезенки;
- гипертоническая болезнь;
- холестериновое кольцо по краю радужки.
Если нарушение липидного обмена привело к снижению количества жиров в организме, то возникают такие симптомы, как выпадение и раннее поседение волос, воспалительные поражения кожи, расслоение ногтей, сбой менструального цикла у женщин.
Особенности лечения
Для коррекции нарушений липидного обмена применяют различные подходы. Если проблема была вызвана алиментарными причинами, то пациента передают в руки диетолога, который разрабатывает план правильного питания и физических упражнений. Если нарушения липидного обмена появились в результате сопутствующих заболеваний, то приступают к их лечению. Некоторые болезни не поддаются терапии (например, сахарный диабет), поэтому врач разрабатывает индивидуальный список рекомендаций, который поможет нормализовать липидный обмен и улучшить качество жизни пациента.
Наследственные болезни обмена липидов
Наследственные болезни обмена липидов — обширная группа заболеваний, возникающих в результате нарушения одной из стадий синтеза, транспорта и деградации липопротеинов, в состав которых входят все основные плазменные липиды — триглицериды (ТГ), фосфолипиды, холестерол (ХС) и свободные жирные кислоты. Липиды плазмы крови наиболее часто находятся в соединении с различными апопротеинами, образуя липопротеины. К настоящему времени лучше всего изучены 8 основных апопротеинов — Аро-А 1,2,4, Аро-В, Аро-С 1,2,3 и Аро-Е. Некоторые апопротеины являются составной частью определенных липопротеинов, другие же могут мигрировать от одного липопротеина к другому.
Существует четыре основных класса липопротеинов плазмы крови:
1) хиломикроны (ХМ) — образуются в клетках слизистой оболочки кишечника и являются носителями пищевых ТГ;
2) липопротеины очень низкой плотности (ЛПОНП) — образуются в печени и используются для экспорта ТГ в другие ткани;
3) липопротеины низкой плотности (ЛПНП) — являются конечными продуктами катаболизма ЛПОНП и переносчиками ХС в клетки;
4) липопротеины высокой плотности (ЛПВП) — осуществляют метаболизм ХМ и ЛПОНП и транспортируют ХС из клеточных мембран в печень.
Типичный липопротеин состоит из липидного ядра, образованного триглицеридами и эфирами холестерола и наружного слоя, представленного фосфолипидами, холестеролом и апопротеинами.
Нормальный метаболизм липидов происходит следующим образом. Синтезированные в кишечнике ХМ и в печени ЛПОНП попадают в кровеносное русло и переносят ТГ (триглицериды) в жировые клетки и другие ткани. После отделения ТГ в жировых клетках, ХМ вновь поступают в печень, а ЛПОНП превращаются в ЛПНП, обогащенные ХС и содержащие Аро-В и Аро-Е. ЛПНП сохраняются в кровеносном русле в течение 2,5 суток и служат транспортерами ХС в различные клетки. Поступление ХС в клетки осуществляется при помощи рецепторов ЛПНП, количество и функции которых находятся под генетическим контролем. Клеточные рецепторы группируются в специальных участках клеточной мембраны, погруженных виде кратера в цитоплазму. После взаимодействия рецептора с ЛПНП участок мембраны, содержащий рецепторы, втягивается внутрь клетки и образует окаймленную везикулу. В цитоплазме ЛПНП отделяются от рецептора и проникают в лизосомы, а рецепторы возвращаются на клеточную мембрану. Освобождение ХС из ЛПНП осуществляется под действием нескольких ферментов. Обратная доставка ХС в клетки печени и тонкого кишечника осуществляется ЛПВП также с использованием механизма рецепторного эндоцитоза.
К настоящему времени описано более 420 различных мутаций, которые в зависимости от воздействия можно сгруппировать в 5 классов:
1) приводящие к прекращению синтеза рецептора;
2) нарушающие миграцию рецептора из эндоплазматического ретикулума в аппарат Гольджи;
3) изменяющие процесс связывания рецептора с ЛПНП;
4) нарушающие кластеризацию рецептора на клеточной мембран;
5) нарушающие процесс отщепления ЛПНП в клетке и транспорт рецептора к мембране клетки.
Основные этапы обмена ХС в организме находятся под сложным генетическим контролем. Нарушение в результате мутаций различных этапов этого метаболического пути может привести к задержке выведения ХС из организма и возникновению различных вариантов гиперлипидемий, развитию атеросклероза сосудов сердца и мозга. В последние годы удалось расшифровать молекулярно-генетические механизмы некоторых моногенных болезней, обусловленных нарушением обмена липидов.
Липоидозы — большая группа наследственных аномалий липидного обмена, общей чертой которых является высокий уровень липидов в плазме крови (плазматические липоидозы) либо накопление метаболитов липидного обмена внутри клеток (внутриклеточные липоидозы). К липоидозам относят болезни обмена липопротеидов. Наиболее распространенными формами липоидозов являются болезни Гоше и Ниманна-Пика, Тей-Сакса, относящиеся к так называемым «болезням накопления», клиническая дифференцировка которых крайне трудна.
Болезнь Гоше относится к сфинголипидозам — болезням накопления липидов; обусловлена дефектом гена, ответственного за синтез лизосомального гидролитического фермента бета — глюкоцереброзидазы (бета — гликозидазы).
Заболевание наследуется по аутосомно-рецессивному типу.
Дефект и дефицит этого фермента ведут к нарушению утилизации липидов — глюкоцереброзидов и накоплению их в макрофагах преимущественно костного мозга, селезенки, печени. Диагноз устанавливают по обнаружению специфических клеток Гоше (лимфоцитоподобное ядро, эксцентрично расположенное, и очень широкая светлая цитоплазма с чуть приметной циркулярной исчерченностью) в пунктате селезенки (пункцию ее можно делать только в стационаре) или в костном мозге.
Выделяют три типа болезни Гоше. Тип 1 (доброкачественный) – взрослый тип. В 30 раз чаще встречается у евреев (западноевропейской группы Ашкенази). Неврологические нарушения при этом отсутствуют, висцеральные изменения связаны преимущественно с кроветворными органами, увеличением селезенки, явлениями гиперспленизма, деструкцией костной ткани.
Тип 2 – инфантильный. Представляет собой злокачественную форму процесса с грубыми неврологическими нарушениями, которые проявляются уже у новорожденных и ведут к смерти в первые 2 года жизни.
Тип 3 – ювенильный. Отличается вариабельностью висцеральных и неврологических нарушений; по течению он менее злокачествен, чем тип 2. Разнообразие форм болезни Гоше обусловлено гетерогенностью мутаций гена бета — гликозидазы.
Бозень Нимана — Пика (сфингомиелиноз). Относится к группе тезауризмозов (болезней накопления) и характеризуется тяжелым поражением нервной системы, задержкой общего развития ребенка.
Наследование болезни происходит по аутосомно-рецессивному типу. Мальчики и девочки болеют одинаково часто. Нередко отмечается кровное родство между родителями, имеются семейные случаи болезни Нимана — Пика.
Патогенез болезни связывается с нарушением синтеза сфингомиелина, в составе которого отмечается избыточное количество одних жирных кислот и недостаток других. Возможно, определенное значение имеют нарушения процессов распада сфингомиелина, что приводит к его избыточному накоплению в ретикуло-эндотелиальных клетках различных органов и тканей. Микроскопически в селезенке, печени, почках, надпочечниках, лимфатических узлах, костном мозге и некоторых других органах обнаруживаются клетки Пика. Это довольно крупные клетки, имеющие размер от 20 до 50 мк и содержащие одно или много ядер. Протоплазма клеток содержит вакуоли, придающие клеткам характерный пенистый вид. Пенистость клеток образуется вследствие растворения липидных субстанций при фиксировании препарата. Гистологические изменения обнаруживаются в ганглиозных клетках головного мозга и сетчатой оболочки глаза. Количество сфингомиелина в различных органах резко увеличивается. В 1961 году, была предложена следующая классификация заболевания:
— болезнь Ниманна-Пика, тип А: классическая инфантильная;
— болезнь Ниманна-Пика, тип В: висцеральная;
— болезнь Ниманна-Пика, тип С: неострая подростковая
— болезнь Ниманна-Пика, тип D: ново-шотландская;
Однако, на сегодня, когда понятна генетическая природа заболевания, расстройство классифицируется следующим образом:
— болезнь Ниманна-Пика, связанная с геном SMPD1, которая включает в себя типы А и В;
— болезнь Ниманна-Пика, типа C, который включает в себя типы C1 и C2. (Тип D возникает в результате мутации того же гена, что и тип C1).
Клинически болезнь Нимана — Пика проявляется обычно в грудном возрасте, чаще в первое полугодие. Имеются единичные описания возникновения клинических признаков болезни в более старшем детском и даже молодом возрасте. Начальными симптомами болезни являются потеря аппетита, резкое похудание ребенка, задержка психофизического развития. Живот больного значительно увеличивается вследствие гепатоспленомегалии (увеличение селезенки и печени). Часто возникают бронхопневмонии. Лимфатические железы в большинстве случаев увеличиваются. Кожные покровы выглядят восковидными, блестящими, обнаруживаются участки пигментации. Неврологически в начальных стадиях болезни выявляются двигательные расстройства центрального характера: парезы конечностей, повышение тонуса мышц и сухожильных рефлексов, пирамидные знаки. В более поздних стадиях характерна гипотония мышц, отсутствие сухожильных рефлексов. Развивается идиотия, слепота и глухота. На глазном дне у многих больных могут обнаруживаться атрофия сосков зрительного нерва, вишнево-красное пятнышко овальной формы в макулярной области. Болезнь Нимана — Пика обладает злокачественным течением. Большинство детей погибают в первые 2 года жизни от легочно-сердечной недостаточности, интеркуррентных инфекций.
Болезнь Тея-Сакса — наследственное нейрометаболическое заболевание, связанное с накоплением в головном мозге ганглиозида GM2. Ганглиозиды содержат один или более остатков сиаловой кислоты.
Аутосомно-рецессивный тип наследования. Болезнь Тея-Сакса обусловлена мутационными поражениями гена гексозаминидазы (HEXA), контролирующего синтез альфа субъединицы гексозаминидазы А. Гексозаминидаза А — лизосомный фермент, катализирующий катаболизм GM2 ганглиозида.
Различают три формы болезни Тея — Сакса:
Детская форма — через полгода после рождения у детей отмечается прогрессирующее ухудшение физических возможностей и умственных способностей: наблюдаются слепота, глухота, потеря способности глотать. В результате атрофии мышц развивается паралич. Смерть наступает в возрасте до 3—4 лет.
Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания) дизартрия, (расстройства речи), атаксия (шаткость походки), спастичность (контрактуры и параличи). Смерть наступает в возрасте до 15—16 лет.
Взрослая форма — возникает в возрасте от 25 до 30 лет. Характеризуется симптомами прогрессирующего ухудшения неврологических функций: нарушение и шаткость походки, расстройства глотания и речи, снижение когнитивных навыков, спастичность, развитие шизофрении в форме психоза.
Болезнь Фабри является редкой наследственной Х-сцепленной рецессивной лизосомальной болезнью накопления. Дефицит фермента альфа-галактозидазы А, который возникает из-за мутации, вызывает накопление гликолипида известного как глоботриаосилцерамид (сокращенно GB3, GL-3) в кровеносных сосудах, других тканях и органах. Такое накопление приводит к нарушению их нормального функционирования.
Боль во всем теле или локализованные боли в конечностях (известный как акропарестезия) или желудочно-кишечном тракте (ЖКТ) является характерным явлением для пациентов с болезнью Фабри. Считается, что акропарестезия при заболевании связана с повреждением периферических нервных волокон, передающих боль. Боль в желудочно-кишечном тракте, вероятно, обусловлена накоплением липидов в малых его сосудах, что в свою очередь, мешает кровообращению (ишемия кишечника), вызывая боль. Распространенным и серьезным следствием болезни являются осложнения, связанные с почками. Почечная недостаточность (уремия) может ухудшаться со временем. Протеинурия (которая может вызвать пенистость мочи) часто является первым признаком поражения почек. Конечная стадия почечной недостаточности у мужчин, как правило, наступает в 30-40 летнем возрасте, и является частой причиной смерти. Ангиокератомы (маленькие, безболезненные папулы, которые могут появляться на любой части тела, но обычно появляются на бедрах, вокруг пупка, ягодицах, нижней части живота и в паху) тоже является распространенным симптомом. Ангидроз (отсутствие потоотделения) является распространенным симптомом в отличие от гипергидроза (повышенная потливость), который встречается значительно реже. При заболевании характерны также поражение глаз, а именно помутнение роговицы (известное как кератопатия).
Наследственный дефицит печеночной липазы,или гипер-a-триглицеридемия, характеризуется накоплением ТГ во фракции a-ЛП. В результате торможения катализируемых печеночной липазой реакций в крови. Проявления: липоидная дуга роговицы, ксантомы, ладонные стрии, ИБС.
Гиперлипидемия — повышение уровня липидов в крови или тканях, обусловленное врожденным или приобретенными нарушением обмена веществ.
Генетические факторы взаимодействуют с факторами окружающей среды, включающими образ питания и прием лекарств. Наследственные компоненты часто являются полигенными, и плохо поддаются определению, но тем не менее описано три чисто наследственных нарушения — семейная гиперхолестеринемия (СГХК), семейная гиперлипопротеинемия III типа и семейная комбинированная гиперлипидемия.
Различают пять типов гиперлипопротеинемий с характерными для каждого типа специфическими симптомами:
Тип I характеризуется приступами боли в желудке, обычно после употребления жирной пищи, а также общим ухудшением самочувствия, потерей аппетита и повышением температуры.
Тип II характеризуется появлением плотных образований на ахилловых сухожилиях и сухожилиях кистей рук и стоп
Тип III может вызывать появление над локтями и коленями мягких воспаленных язвочек. Тип IV обусловлен перееданием, ожирением и диабетом.
Тип V проявляется болями в животе (самый распространенный симптом), желтыми узелками на коже и красновато-беловатыми сосудиками на сетчатке глаз
Наследственная гиперхолестеринемия – аутосомно-доминантное, моногенное заболевание, причиной развития которого является нарушение структуры или функции рецепторов к липопротеинам низкой плотности (ЛПНП) на соматических клетках (печёночных и других) и/или их количества [5,13] (классическая форма НГХС [5]), или нарушения в структуре молекулы аполипопротеинов В-100 и С (апо В-100 и С, белковых компонентов липопротеинов).
В зависимости от природы генетического дефекта это заболевание классифицируется как рецептор-негативная, рецептор-дефективная или рецептор-интернационализационная формы.
Основными нарушениями являются нарушенный катаболизм ЛПНП печенью и другими тканями через ЛПНП-рецепторы, которые связываются с Apo В. При патологии ЛПНП-рецепторов это вызывает повышение уровня ЛПНП в крови и увеличение времени циркуляции данного вида липопротеинов, что в конце концов ведёт к образованию изменённых, модифицированных форм ЛПНП, обладающих высокой атерогенностью. Гомозиготные формы НГХС отличаются от гетерозиготных полным отсутствием ЛПНП-рецепторов на поверхности клеток и удаления ЛПНП из кровотока данным путём, и более длительной циркуляцией их периферическом кровяном русле в результате данных изменений (захват ЛПНП клетками происходит нерецепторным способом), тогда как при гетерозиготных формах число рецепторов снижено примерно на половину.
На коже больных НГХС отмечаются патогноматические признаки нарушения липидного обмена: ксантомы и ксантелазмы (отложение эфиров ХС в толще кожи).
Методы диагностики
Перед составлением плана обследования врач беседует с пациентом, определяет особенности его питания, выясняет, принимает ли он какие-либо лекарственные препараты, были ли случаи нарушения липидного обмена у его родственников. После этого назначается комплексное обследование, в которое могут входить следующие методы:
- общий и биохимический анализ крови;
- липидограмма (определение уровня холестерина, липопротеинов высокой и низкой плотности);
- УЗИ внутренних органов;
- определение уровня гормонов;
- антропометрия;
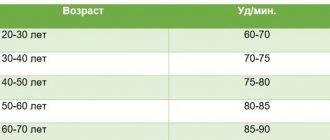
- ЭКГ;
- офтальмологический осмотр и др.
С целью выявления дефектных генов, которые могут приводить к нарушению липидного обмена, назначается генетическое тестирование. Пройти его можно в медико-генетическом .