Талассемия — это не одна, а группа похожих болезней, передающихся по наследству и вызванных нарушенным синтезом белковой части гемоглобина. Заболевание достается ребенку с вероятностью в 100%, если один из родителей содержит измененный ген. Поэтому оно считается самой распространенной наследственной патологией.
Наибольшее число случаев зарегистрировано в странах, расположенных по берегам Средиземного моря, на африканском континенте, в Средней Азии, Индии. Среди населения Азербайджана каждый десятый имеет измененные гены.
Немного истории
Болезнь была впервые описана в 1925 году американскими врачами-педиатрами, которые при лечении эмигрантов из Италии выявили одинаковую клиническую картину у детей с тяжелым малокровием, увеличением печени и селезенки, изменениями костей.
Затем появились работы, описывающие более легкое течение болезни у взрослых пациентов. Термин «талассемия» предложили в 1936 году. Дословно он означает «болезнь морского побережья». Высказана мысль о связи патогенеза заболевания с нарушением синтеза глобиновых цепочек.
Почему разрушаются эритроциты?
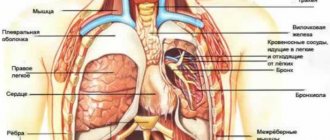
Патогенез талассемии достаточно изучен. Первичные изменения начинаются с нарушенного синтеза гемоглобина, а именно белковых цепочек вещества. Известно, что гемоглобин, входящий в состав 90% массы эритроцитов, — единственное вещество, способное связывать молекулы кислорода и разносить их из легочной ткани по всему организму.
Его структура состоит из пигмента (гема), включающего железо, и набора из двух пар белковых цепочек. Их называют по типичному расположению аминокислот альфа- и бета-цепями. При нарушенном синтезе одного из типов полипептидов накапливается другой вид.
В результате разрушаются все клетки эритроцитарного ряда (сами эритроциты и их предшественники), из них выходит гемоглобин. Малокровие (анемия) развивается по гипохромному типу. Это подтверждается низким цветовым показателем.
«Виновниками» разрушений являются гены, ответственные за построение белковой части гемоглобина. Их мутация нарушает способность составлять необходимый набор аминокислот в цепи. Причину изменений связывают с возбудителем малярии — плазмодием. Доказаны его мутирующие способности. Талассемия по территории распространенности совпадает с эпидемическими зонами малярии.
Основные четыре цепочки белков, связывающие гем и влияющие на эритроциты
Дети могут получить болезнь путем наследования от родителей в двух видах:
- гомозиготном — ген-мутант передается от обоих родителей;
- гетерозиготном — ген болезни передается только от матери или отца, носителем может быть один из родителей.
Соответственно, называются формы талассемии.
При гетерозиготном носительстве выделяют:
- «немой» ген (α-th2);
- «манифестный» ген (α-th1).
Наследование и эпидемиология
При этом в мире всего пять процентов населения – носители мутаций в генах цепей гемоглобина, а из них симптомам болезни подвержены чуть менее двух процентов людей. Стоит также учитывать, что жители разных регионов сталкиваются с талассемией с разной частотой. Прежде всего болезнь поражает жителей Средиземноморья, а также стран Центральной и Южной Азии. В России носителей мутаций, связанных с талассемией, сравнительно мало – около одного процента (для бета-типа).
Носители мутации могут даже не подозревать о своем статусе – часто повреждение лишь одной копии гена не ведёт к тяжелым симптомам. Однако носители мутации рискуют передать болезнь детям: если два носителя заведут ребенка, он может получить от родителей сразу обе копии мутировавшего гена с вероятностью около 25 процентов.
В предыдущем разделе мы уже обсуждали, что мутации в двух копиях генов приводит к более тяжелой форме талассемии. Но если посчитать вероятность такого события получится лишь около сотой процента.
Разновидности заболевания
Все виды заболевания связаны с кислородной недостаточностью, наступившей вследствие разрушения эритроцитов — единственных клеток, обеспечивающих газообмен в органах и тканях.
В зависимости от сбоя синтеза полипептидной цепочки различают:
- альфа-талассемию;
- бета-талассемию.
Наиболее распространена бета-талассемия. Она характеризуется избыточным накоплением α-цепочек глобина.
Выявлены также редкие формы болезни, которые названы гамма- и дельта-талассемией. В группу включены:
- некоторые гемоглобинопатии;
- гомозиготная альфа-талассемия с водянкой плода;
- смешанный вид β- и δ-талассемии, когда поражаются одновременно дельта- и бета-цепи.
Каждая форма имеет свои типичные клинические проявления и отличия. Но в зависимости от тяжести течения принято выделять:
- хроническую легкую — люди доживают до старости;
- хроническую среднетяжелую — пациенты не переживают детский возраст;
- тяжелую — ребенок погибает в период новорожденности.
Признаки и симптомы
Представления людей с альфа-талассемией включают:
Признаки и симптомы альфа-талассемии[8][9][10]
| Общий | Необычный |
|
|
Клинические проявления большой талассемии
Симптомы талассемии зависят по клинике и времени проявления от вида генной мутации.
Большая талассемия (болезнь Кули) развивается при гомозиготной передаче от родителей. Ее проявления заметны у детей сразу после рождения.
У новорожденного замечают:
- удлиненный вверх череп («башенный»);
- более развитую верхнюю челюсть;
- монголоидный тип лицевого скелета.
Типичная внешность и изменения структуры костей черепа используются в диагностике
К 12-месячному возрасту проявляется:
- расширенная носовая перегородка, «приплюснутый» нос;
- костные наросты на стопах;
- нарушенный прикус;
- желтушность кожи (в связи с поражением селезенки).
Кислородное голодание тканей приводит к органным нарушениям. При осмотре отмечаются:
- увеличение печени, край плотный на ощупь (развивается ранний цирроз);
- возникновение сердечной недостаточности (лишнее железо откладывается в миокарде и поражает сократимость сердечной мышцы);
- отставание в физическом и умственном развитии от сверстников.
У малыша развивается сахарный диабет из-за фиброзирования поджелудочной железы и печеночная недостаточность. В тяжелых случаях он живет не более одного года.
В старшем возрасте проявляются такие осложнения:
- трофические язвы на коже, вызванные локальными нарушениями кровообращения;
- частые переломы костей;
- повторные воспаления в легких;
- калькулезный холецистит;
- кардиосклероз;
- сепсис при контакте с инфекцией;
- нарушенное полового развитие.
Выделяют промежуточную форму β-талассемии с более доброкачественным течением. Она начинается у детей старшего возраста. Внешний вид ребенка не изменяется. Нет отставания в умственном и физическом развитии. Основные проявления вызываются увеличением селезенки и повышенной ломкостью костной ткани.
Патофизиология
Механизм показывает, что альфа-талассемия приводит к снижению продукции альфа-глобина, следовательно, образуется меньше цепей альфа-глобина, что приводит к избытку β-цепей у взрослых и избытку γ-цепей у новорожденных. Избыточные β-цепи образуют нестабильные тетрамеры, называемые гемоглобин H или HbH четырех бета-цепочки. Избыточные γ-цепи образуют тетрамеры, которые являются плохими переносчиками O2 поскольку их близость к O2 слишком высока, поэтому не диссоциирует на периферии. Гомозигота α0 талассемии, где многочисленные γ4 но никакие α-глобины не встречаются вообще (называемые Hb Barts), часто приводят к смерти вскоре после рождения.[3][1][13]
Клиника малой талассемии
Малая или гетерозиготная форма передается одним из родителей. Второй «здоровый» ген сглаживает повреждения. Симптомы талассемии могут длительно не проявляться или вызывать признаки, общие с другими заболеваниями:
- слабость, повышенную утомляемость;
- головные боли;
- головокружение.
При осмотре обращается внимание на следующее:
- бледность кожи с желтушным оттенком;
- возможно увеличение печени и селезенки.
У ребенка с малой формой резко увеличена восприимчивость к инфекциям, снижен иммунитет. Очень тяжело и часто протекают вирусные и кишечные инфекционные заболевания.
Во взрослом состоянии у женщины при беременности развивается водянка плода (повышение внутричерепного давления и скопление жидкости в желудочках мозга). Эта патология несовместима с жизнью ребенка, в случае нормальных родов ведет к тяжелым неврологическим и психическим отклонениям.
Минимальной β-талассемией называют синдром Сильвестрони — Бьянко. Эта форма заболевания протекает практически бессимптомно, обнаруживается случайно в семьях со случаями талассемии.
Диагностика
Диагностика талассемии должна начинаться с генетической консультации супругов перед зачатием ребенка. Во время беременности при необходимости проводят анализ амниотической жидкости. Уже на ранних сроках в эритроцитах плода можно обнаружить характерные изменения.
Порой внешний осмотр и выяснение семейного анамнеза позволяют заподозрить болезнь у ребенка без лабораторной диагностики.
В анализе крови выявляют:
- снижение уровня гемоглобина до 30–50 г/л при гомозиготном варианте и до 90 –110 г/л при гетерозиготном;
- низкий цветовой показатель (менее 0,5), который образуется малым насыщением клеток гемоглобином);
- рост ретикулоцитов (предшественников эритроцитов) до 4%.
При просмотре окрашенного мазка под микроскопом обращают внимание на следующее:
- наличие слабо окрашенных (гипохромных) эритроцитов;
- изменение размеров и формы красных клеток, эритроциты приобретают формы овала, серпа, шара.
Одна из характерных разновидностей серповидных клеток — дрепаноциты.
Дрепаноциты образуются при любых гемоглобинопатиях, содержат особую разновидность (S гемоглобин), который может в условиях низкой концентрации образовывать полимеры и изменять форму оболочки клетки
Биохимические тесты показывают нарушенный обмен железа, как при гемолитической анемии:
- высокий уровень свободного билирубина;
- повышенную концентрацию железа в сыворотке;
- сниженную способность к связыванию железа.
Исследование особенностей гемоглобина проводят с помощью ацетат-целлюлозной пленки. Подобный анализ позволяет определить количественный уровень фракций. Гомозиготная бета-талассемия отличается увеличенным уровнем фетального гемоглобина (в норме он содержится у плода, а у взрослого человека всего до 1%), неспособного переносить кислород.
Использование метода полимеразной цепной реакции позволяет изучить строение полипептидных цепочек гемоглобина.
Генетический анализ выявляет мутацию в одиннадцатой паре хромосом при β-талассемии, другие специфические изменения, типичные для прочих форм.
Исследование пунктата костного мозга проводится для выявления повышенного содержания незрелых эритроцитов в виде сидеробластов.
Рентгенологическое исследование способствует обнаружению дефектов костной ткани:
- участков остеопороза (сниженной плотности);
- увеличенной массы костей черепа;
- поперечной исчерченности на мелких костях кистей и стоп;
- на рентгенограмме черепа при большой форме β-талассемии видно типичное игольчатое поражение надкостницы, которое называется симптомом «волосатого черепа».
Ультразвуковое исследование подтверждает увеличение печени и селезенки, помогает обнаружить камни в желчевыводящей системе.
Лабораторные данные более отчетливо выражены при β-талассемии. Другие формы болезни не дают четкой картины.
Заболевания, с которыми необходимо проводить дифференциальную диагностику:
- железодефицитная анемия,
- серповидно-клеточная анемия,
- гемолитическая аутоиммунная анемия,
- наследственный микросфероцитоз.
Анемии, патология гемостаза, онкогематология
Материалы представлены из учебного пособия РУДН
Анемии. Клиника, диагностика и лечение / Стуклов Н.И., Альпидовский В.К., Огурцов П.П. – М.: ООО «Медицинское информационное агентство», 2013. – 264 с.
Копирование и тиражирование материалов без указания авторов запрещено и преследуется по закону.
Талассемии – это группа наследственных заболеваний, относящихся к количественным гемоглобинопатиям, при которых имеет место нарушение синтеза полипептидных цепей глобина β или α, что приводит к уменьшению образования одной или нескольких фракций нормального гемоглобина или, в редких случаях, – формированию гемоглобина, не встречающегося у здоровых людей.
При гомозиготных состояниях этот дефект синтеза цепей глобина проявляется гемолитической анемией разной степени выраженности. При гетерозиготных β-талассемиях, когда патологический ген наследуется от одного из родителей, а также у некоторых больных с гомозиготной α-талассемией заболевание проявляется слабовыраженной гипохромной анемией, а признаки гемолиза отсутствуют.
Впервые талассемия была описана Cooley и Lee в 1925 году при наблюдении 5 детей из итальянской семьи. У всех детей в семье наблюдалась гипохромная гемолитическая анемия со значительным увеличением селезенки и печени, изменения костей скелета. Заболевание назвали болезнью Кули. Позднее были описаны более легкие формы, и в 1936 году болезнь получила название «талассемия».
В зависимости от нарушения синтеза α- или β-цепей глобина выделяют две основные формы заболевания: α- и β-талассемию.
β-талассемии
β-талассемия обусловлена рядом мутаций в локусе β-глобина на 11 хромосоме, нарушающих синтез β-цепей. Эти мутации могут происходить на разных стадиях экспрессии гена, отвечающего за синтез β-цепей: дефект ДНК, нарушение гена-регулятора цепей глобина или транспортной РНК. При ограничении транскрипции т-РНК обычно снижается синтез β-цепей – β+-талассемия. При β0-талассемии мутация происходит в зоне кодирования и вызывает полную остановку синтеза β-цепей. Всего описано более 100 различных мутаций локуса β-глобина, которые передаются доминантно, но иногда возникают в результате спонтанных мутаций.
Патогенез клинических проявлений при гомозиготной β-талассемии связан не только с недостаточной продукцией нормального гемоглобина, но и с относительной избыточной выработкой малорастворимых α-цепей, свободные α-цепи образуют тетрамеры. В количественном отношении α-цепи вырабатываются нормально. Эти избыточные α-цепи в виде внутриклеточных преципитатов выпадают в осадок, разрушая мембрану клеток-предшественников эритроцитов, вызывая их внутрикостномозговую гибель (неэффективный эритропоэз) и гемолиз уже образовавшихся эритроцитов. Все это приводит к развитию основного симптома гомозиготной талассемии – анемии.
В ответ на развитие тканевой гипоксии, сопровождающей анемию, почки значительно повышают выработку эритропоэтина, который стимулирует формирование кроветворной ткани в костях, где она обычно отсутствует, вызывая деформацию костей (башенный череп, монголоидность и др.), а также образование экстрамедуллярного кроветворения в печени и селезенке с их выраженным увеличением.
β-талассемия широко распространена в странах Средиземного моря (Италия, Греция, Кипр, Турция и др.), Центральной и Восточной Африке, Ближнем и Среднем Востоке, Индии, Юго-Восточной Африке. Широкая распространенность заболевания, предполагается, является результатом лучшей переносимости малярии больными-гетерозиготами по β-талассемии – механизм остается неясным.
Выраженность клинических проявлений при гомозиготной β -талассемии зависит от степени нарушения синтеза β-цепей.
Самая тяжелая форма, или болезнь Кули, обозначается как большая β0-талассемия. При этой форме полностью отсутствует синтез β-цепей.
Клиника
Анемия при большой талассемии развивается уже к первому году жизни ребенка и сопровождается отставанием в психическом и физическом развитии. Со временем анемия прогрессирует, появляется желтушность кожи и склер, значительно увеличиваются селезенка и печень, нередко на коже ног образуются трофические язвы. Характерен внешний вид больных детей с болезнью Кули: непропорционально большая деформированная голова («башенный череп»), приплюснутый нос, суженые глазные щели и выпуклые скулы (признаки «монголоидности»). Дети резко отстают в развитии, отмечается повышенная восприимчивость к инфекциям, половое недоразвитие.
При рентгенологическом исследовании трубчатых костей отмечается истончение кортикального слоя и расширение костномозгового канала. Для рентгенографической картины костей черепа характерно расширение диплоического пространства костей свода черепа и появление радиальной исчерченности – вид «ёжика» или «щетки». Сходные изменения структуры наблюдаются в метафизах трубчатых костей, плоских костях таза, лопатках и ребрах.
В анализах крови – глубокая анемия, концентрация гемоглобина – 30-50 г/л, выраженная гипохромия, анизоцитоз, значительная мишеневидность эритроцитов, базофильная пунктация. При электрофорезе гемоглобина эритроцитов 98% составляет HbF, 2-3% – HbA2 и большое количество свободных α-цепей.
Ранее прогноз при этой форме заболевания был крайне неблагоприятным, поскольку большинство детей погибало в возрасте 3-4 лет. В настоящее время выздоровление детей с той тяжелой патологией возможно после проведения трансплантации аллогенного костного мозга.
При среднетяжелой и промежуточной форме β+-талассемии смягчение тяжести заболевания обусловлено частичным сохранением синтеза β-цепей, а, следовательно, присутствием в эритроцитах нормального HbA.
От способности синтезировать β-цепи зависит тяжесть течения заболевания. При незначительной продукции β-цепей эта форма β+-талассемии, тип I, обозначается как среднетяжелая и по клинике приближается к болезни Кули. Больные доживают до школьного возраста, а в их эритроцитах преобладает HbF 60 — 80%, HbA2 3-9%, свободные α-цепи и появляется небольшое количество HbA.
Больные с β+-талассемией, тип II, с более выраженной продукцией β-цепей, описывается как промежуточная форма (thalassemiaintermedia), с менее выраженной анемией. Больные доживают до зрелого возраста, при адекватной гемотрансфузионной терапии практически не отстают в развитии и способны заканчивать школы, колледжи и даже университеты.
Диагностика гомозиготной β-талассемии не вызывает больших трудностей:
— признаки гемолитической анемии с детства;
— отставание в развитии и деформация скелета (болезнь Кули и среднетяжелая форма);
— гепато-, спленомегалия;
— мишеневидность и гипохромия эритроцитов;
— преобладание при электрофорезе гемоглобина HbF и свободных α-цепей, увеличение А2 Hb;
— молекулярная диагностика (исследование мутаций генов, кодирующих цепи Hb).
Лечение
Большим достижением в лечении тяжелой и среднетяжелой форм β-талассемии стало применение аллогенной трансплантации костного мозга (чаще от брата или сестры), позволившее в 80 – 90% добиться выздоровления ранее некурабельных больных.
Больные с промежуточной формой β+-талассемии, как и больные с тяжелой формой заболевания, не могут жить без периодических трансфузий эритроцитарной массы, частота которых регулируется уровнем концентрации гемоглобина.
Для нормального развития ребенка рекомендуется поддерживать концентрацию гемоглобина на уровне 100 – 110 г/л. Регулярные гемотрансфузии, в сочетании с повышенным всасыванием железа в кишечнике у анемизированных больных создает опасность развития гемосидероза с поражением сердца (кардиомегалия), печени (фиброз), поджелудочной железы (сахарный диабет), почек (почечная недостаточность) и других органов. Поэтому гемотрансфузионная терапия должна сочетаться с применением хелатирующих лекарственных препаратов (десферал, эксиджад), которые способны связывать свободное сывороточное железо, внутриклеточное железо из гепатоцитов, извлекать его из комплекса с трансферрином и ферритином.
Десферал вводится внутривенно, длительно, по 1-2 г препарата на каждые 500 мл донорской эритроцитарной массы. Возможно длительное подкожное введение с помощью инжектора с одновременным определением сывороточного ферритина.
В последние годы разработан препарат со свойствами десферала – эксиджад (деферазирокс), обладающий выраженным преимуществом, поскольку он может применяться внутрь, по 20 мг/кг массы тела больного.
При массивной спленомегалии рекомендуется проведение спленэктомии. Показания:
— прогрессирующе увеличение размеров селезенки (>8 см из-под края реберной дуги),
— увеличение потребности в гемотрансфузиях более, чем на 50% от исходного уровня в течение 6 месяцев,
— при повышении потребности в эритроцитарной массе более, чем на 250 мл в течение года.
α-талассемии
α-талассемия впервые была описана в 1955 году. Выявляется в Греции, Таиланде, Нигерии, Азербайджане, Дагестане. В основе α-талассемии лежит нарушение синтеза α-цепей глобина. Поскольку эти цепи входят в состав всех нормальных фракций гемоглобина, при α-талассемии происходит равномерное снижение их синтеза.
Две почти идентичные копии гена α-глобина находятся на хромосоме 16. Наиболее часто встречается потеря одного или нескольких их этих 4 генов – 80-85% случаев α-талассемии.
Клинические проявления α-талассемии напрямую коррелируют со степенью нарушения синтеза α-глобиновых цепей. При данном виде аномалии симптомы менее выражены, чем при β-талассемии, что связано с наличием 4 генов α-глобина. Адекватное количество α-цепей образуется до тех пор, пока не утрачиваются 3 или 4 гена. Кроме того, β4-тетрамер (или HbH), образующийся при недостатке α-цепей, более растворим, чем тетрамер α4. Вследствие этого даже при выраженном нарушении синтеза α-цепей при α-талассемии гемолиз гораздо менее выражен, а эритропоэз более эффективен, чем при β-талассемии.
Гемоглобин Барт – гомозиготная α-талассемия I – поражены все 4 гена, ответственные за синтез α-цепей – самая тяжелая форма α-талассемии. Встречается такая форма только в странах Юго-Восточной Азии. Это состояние не совместимо с жизнью. Беременность в подобных случаях заканчивается самопроизвольным выкидышем, мертворождением или гибелью плода в первые часы жизни (водянка плода), так как у плода не синтезируется фетальный гемоглобин (α2γ2). Свободные γ-цепи образуют тетрамеры (γ4), так называемый гемоглобин Барт. Такой гемоглобин обладает высоким сродством к кислороду, что приводит к гипоксии тканей плода, сердечной недостаточности, и, в итоге, к гибели плода. В крови – выраженные эритробластемия, гипохромия, макроцитоз, мишеневидность, анизо- и пойкилоцитоз. Электрофоретически в эритроцитах обнаруживается Hb Барт (80-90%) в сочетании с HbH.
H-гемоглобинопатия
Н-гемоглобинопатия вызвана утратой или дисфункцией трех α-глобиновых генов. Гемоглобин Н представляет собой тетрамер β4, образующийся при избытке β-цепей. Клинически близок к промежуточной форме β-талассемии.
Заболевание проявляется к концу первого года жизни гемолитической анемией различной степени выраженности. Характеризуется нетяжелым течением, значительным увеличением селезенки и печени. Отмечается слабо выраженная желтуха из-за увеличения непрямого билирубина. Концентрация гемоглобина – в пределах 70 – 80 г/л, выявляется гипохромия, мишеневидность и базофильная пунктация эритроцитов.
На фоне приема лекарственных препаратов или интеркуррентной инфекции могут развиваться гемолитические кризы с падением уровня гемоглобина до 50 г/л. При электрофорезе доля HbH составляет 5-30% при нормальном уровне HbF и снижении HbA2.
Лечение практически не отличается от лечения гетерозиготной β-талассемии.
Генетическая интерпретация видов бета-талассемии
Ученые-генетики выяснили интересную закономерность: люди имеют одинаковую мутацию генов, отвечающих за синтез гемоглобина, но клиника и степень выраженности заболевания у них отличаются.
Гены могут находиться в следующих состояниях:
- нормальные — характерны для здорового человека;
- поврежденные частично — «работает» неполно, из-за чего синтез полипептидных цепей недостаточен;
- разрушенные полностью — синтез останавливается.
По этому признаку виды талассемии делят на:
- минор — самая легкая форма, поврежден только один ген, внешне человек выглядит здоровым, по анализу крови можно предположить небольшую анемию;
- интермедиа — недостаток бета-цепей серьезно сказывается на синтезе, эритроциты недоразвиты, малокровие выражено с явными признаками, но организм еще может приспособиться, поэтому отсутствует необходимость в постоянных переливаниях крови;
- майор — мутации подверглись все гены, больному необходимы постоянные переливания крови по жизненным показаниям.
Частота встречаемости
Талассемия наследуется аутосомно-рецессивно. Это значит, что поражает одинаково и мальчиков, и девочек. Рецессивный характер говорит о том, что дети больные талассемией появляются в тех семьях, где мама и папа оба являются носителями мутаций. Хотя они могут даже не догадываться о своём носительстве, так как симптомы порой бывают незаметны.
В среднем частота встречаемости составляет 1 на 100 000 человек и может изменяться в зависимости от региона. Бета-талассемия встречается чаще, чем альфа.
Генетические разновидности альфа-талассемии
По охвату мутацией генов, их локусов (определенных частей, отрезков), отвечающих за синтез альфа-цепочек полипептидов, предлагается выделение нескольких групп заболевания:
- мутация только в одном локусе — клинических проявлений нет;
- изменения двух (пары) локусов в одном или разных генах — анализ крови показывает снижение гемоглобина, уменьшение размера эритроцитов;
- поражение трех локусов — выражается в кислородной гипоксии органов, увеличении селезенки, возникновении Н гемоглобинопатии, образующийся гемоглобин нестоек, разлагаясь, вызывает гемолитическую анемию;
- мутация всех локусов — полностью прекращает синтез альфа-цепей, при подобной ситуации происходит внутриутробная гибель плода или ребенок умирает сразу после родов.
Генетические исследования подтвердили также неодинаковое значение пар генов, одна пара из двух является главной, а другая имеет второстепенную роль. Клинические проявления зависят от того, в какой паре произошла мутация.
Пересадка костного мозга считается наиболее результативным способом
Ссылки [ править ]
- ^ abcdefgh
Орига, Рафаэлла; Мои, Паоло; Галанелло, Ренцо; Цао, Антонио (1 января 1993 г.). «Альфа-талассемия» .
GeneReviews
. PMID 20301608 . Проверено 22 сентября 2016 года .обновление 2013 - ^ abПатология BRS
(4-е изд.). Lippincott Williams & Wilkins medical. Декабрь 2009 г. с. 162. ISBN. 978-1451115871 . - ^ abcde
«Обследование альфа-талассемии: подходы, лабораторные исследования, электрофорез гемоглобина» .
emedicine.medscape.com
. Дата обращения 24 мая 2016 . - ^ ab
«Осложнения и лечение | Талассемия | Заболевания крови | NCBDDD | CDC» .
www.cdc.gov
. Проверено 22 сентября 2016 года . - Интернет-Менделирующее наследование в человеке (OMIM): гемоглобин — альфа-локус 1; HBA1 — 141800
- Интернет-Менделирующее наследование в человеке (OMIM): гемоглобин — альфа-локус 2; HBA2 — 141850
- Руководство Ланцковского по детской гематологии и онкологии 6-е издание (2016)
. - ^ abcde
«Альфа-талассемия — Симптомы, диагностика и лечение | BMJ Best Practice» .
bestpractice.bmj.com
. Дата обращения 17 ноября 2022 . - Ссылка, Genetics Home. «Альфа-талассемия» . Домашний справочник по генетике
. Проверено 25 ноября 2022 года . - Орига, Рафаэлла; Мои, Паоло (1993), Адам, Маргарет П .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Альфа-талассемия» , GeneReviews®
, Вашингтонский университет, Сиэтл, PMID 20301608 , получено 25 ноября 2022 г. - «Оценка анемии — Этиология | BMJ Best Practice» . bestpractice.bmj.com
. Проверено 25 ноября 2022 года . - Steensma DP, Gibbons RJ, Хиггс DR (январь 2005). «Приобретенная альфа-талассемия в сочетании с миелодиспластическим синдромом и другими гематологическими злокачественными новообразованиями» . Кровь
.
105
(2): 443–52. DOI : 10.1182 / кровь-2004-07-2792 . PMID 15358626 . - ^ аб
Галанелло Р., Цао А (февраль 2011 г.). «Обзор генного теста. Альфа-талассемия» .
Генетика в медицине
.
13
(2): 83–8. DOI : 10.1097 / GIM.0b013e3181fcb468 . PMID 21381239 . - «Болезнь гемоглобина H» . Сиротка
. Проверено 22 сентября 2016 года . - Vichinsky EP (1 января 2009). «Большая альфа-талассемия — новые мутации, внутриутробное ведение и исходы». Гематология.Американское общество гематологии.Образовательная программа
.
2009
(1): 35–41. DOI : 10,1182 / asheducation-2009.1.35 . PMID 20008180 . - ^ абв
Випракасит, VIP; Экваттанакит, Супачай (1 апреля 2022 г.). «Клиническая классификация, скрининг и диагностика талассемии».
Гематологические / онкологические клиники Северной Америки
. Талассемия.
32
(2): 193–211. DOI : 10.1016 / j.hoc.2017.11.006 . ISSN 0889-8588 . PMID 29458726 . - «UpToDate» . www.uptodate.com
. Проверено 25 ноября 2022 года . - ^ аб
Тахер, Али; Мусаллам, Халед; Каппеллини, Мария Доменика, ред. (2017).
Руководство по ведению нетрансфузионно-зависимой талассемии (NTDT)
(2-е изд.). Международный фонд талассемии. С. 24–32 . Дата обращения 5 ноября 2022 . - «Талассемия | Врач | Пациент» . Пациент
. Проверено 22 сентября 2016 года . - Крегер Э.М., Зингер С.Т., Витт Р.Г., Свитерс Н., Лианоглу Б., Лал А. и др. (Декабрь 2016 г.). «Благоприятные исходы после внутриутробного переливания у плодов с большой альфа-талассемией: серия случаев и обзор литературы». Пренатальная диагностика
.
36
(13): 1242–1249. DOI : 10.1002 / pd.4966 . PMID 27862048 . - Harteveld CL, Хиггс DR (май 2010). «Альфа-талассемия» . Журнал «Орфанет редких болезней»
.
5
(1): 13. DOI : 10,1186 / 1750-1172-5-13 . PMC 2887799 . PMID 20507641 . - Гематология стала проще. АвторДом. 2013-02-06. ISBN 9781477246511. Стр. 246
Проблемы терапии
Лечение талассемии зависит от тяжести поражения эритропоэза, степени охвата генов процессами мутации. В настоящее время применяются следующие методы.
Диетический рацион направлен на снижение всасывания железа в кишечнике, рекомендуются орехи, какао, соя, чай.
Тяжелая форма требует регулярного переливания крови, эритроцитарной массы, размороженных и профильтрованных эритроцитов. Эффективность временная, возможны побочные эффекты, но главное — сохранить жизнь больному.
Терапия дополняется ежедневным устранением излишков железа с помощью введения (хелатов — специальных комплексов, повышающих действие лекарства). Для связывания железа назначается Десферал. Этот препарат предупреждает сидероз (патологическое состояние, вызванное отложением в тканях железа), но на уровень гемоглобина не влияет.
При возникновении резкого ухудшения состояния по аналогии с гемолитическими кризами показаны глюкокортикоиды в больших дозах.
Спленэктомия возможна при больших размерах селезенки детям после пятилетнего возраста. Наиболее оптимален возраст 8–10 лет. После удаления селезенки наступает период улучшения, но опасен риск присоединения инфекции.
Для трансплантации необходим донор, совпадающий по всем параметрам, лучше всего из близких родственников.
К симптоматическим средствам относятся препараты гепатопротекторного действия, большие дозы аскорбинки помогают вывести излишки железа из организма.
Все формы талассемии нуждаются в препаратах с фолиевой кислотой и витаминами группы В. На фоне присоединившейся инфекции, при беременности следует применять фолиевую кислоту в больших дозах, поскольку неэффективное кроветворения при талассемии значительно увеличивает ее потребление клетками.
Уход
Лечение альфа-талассемии может включать: переливание крови для поддержания гемоглобина на уровне, уменьшающем симптомы анемии. Решение о начале переливания зависит от клинической тяжести заболевания.[17]Спленэктомия является возможным вариантом лечения для повышения уровня общего гемоглобина в случаях обострения анемии из-за сверхактивной или увеличенной селезенки или когда трансфузионная терапия невозможна.[18] Однако спленэктомию следует избегать, когда доступны другие варианты, из-за повышенного риска серьезных инфекций и тромбоз.[18]
Кроме того, камни в желчном пузыре может возникнуть проблема, которая потребует хирургического вмешательства. Вторичные осложнения от фебрильный следует контролировать эпизод, и большинство людей живут без лечения.[1][4]
Кроме того, трансплантация стволовых клеток следует рассматривать как лечение (и лекарство), которое лучше всего проводить в раннем возрасте. Другие варианты, например генная терапия, все еще разрабатываются.[19]
Исследование Kreger et al, объединяющее ретроспективный обзор трех случаев большой альфа-талассемии и обзор литературы 17 случаев, показало, что внутриутробное переливание крови может привести к благоприятным результатам. В конечном итоге успешная трансплантация гемопоэтических клеток была проведена у четырех пациентов.[20]