Талассемия – это генетическая болезнь крови, при которой, из-за мутации генов, образовывается недостаточное количество гемоглобина в организме и происходит деформация эритроцитов.
Гемоглобин – это специфический белок, который содержится в эритроцитах и отвечает за перенос железа и кислорода ко всем органам и тканям. Основная функция гемоглобина – дыхательная и его недостаток приводит к кислородному голоданию. В результате чего органы не могут полноценно выполнять свои физиологические функции.
Различают «взрослый» гемоглобин (HbA) и плодный тип гемоглобина (HbF).
Фетальный (плодный) гемоглобин синтезируется у плода спустя 2 недели после формирования внутренних органов (приблизительно с 12-й недели беременности). После рождения ребенка, HbF составляет до 80% и постепенно замещается «взрослым» HbA.
В норме у человека, плодного типа гемоглобина должно остаться не более 1,5%. Если случился сдвиг цифр влево или вправо – это всегда говорит о патологии (может быть нарушена работа сердечно-сосудистой системы, щитовидной железы, гипофиза, наличие гематологических отклонений, лейкоза, лимфогранулематоза, интоксикации).
Здоровая зрелая молекула гемоглобина (HbA) состоит из двух пар цепей – альфы и беты. Фетальный гемоглобин (HbF) состоит из цепей – гаммы и дельты.
А талассемия – это результат нарушенного синтеза одной или нескольких цепей гемоглобина. На основе чего различают альфа-талассемию, бета-талассемию, бета-дельту талассемию и т.д.
Тяжесть протекания того или другого вида заболевания зависит от количества поврежденных участков:
- Если изменен один ген цепи – заболевание (малая талассемия), протекает легко и бессимптомно. Но, при этом человек остается носителем данной болезни, которую может передать по наследству своим детям.
- Нарушение синтеза 2-х генов проявляется микроцитарной анемией в легкой степени тяжести.
- Патология 3-х генов протекает тяжело с ярко выраженной симптоматикой.
- Повреждение сразу 4-х генов (большая талассемия) – это наиболее редкий вид талассемии, который практически несовместим с жизнью (иногда ситуация решается пересадкой костного мозга).
Большая альфа-талассемия – самое серьезное отклонение, которое возникает еще в период внутриутробного развития плода. Грозит осложнениями, как для ребенка, так и для женщины.
Сегодня при своевременной внутриутробной трансфузии эритроцитарной массы удается сохранить жизнь плода.
Большая бета-талассемия (анемия Кули) – это также опасное нарушение, которое проявляется в первые два года жизни ребенка.
Малая альфа и бета-талассемия нарушает процесс эритропоэза (развитие новых эритроцитов), что приводит к хронической анемии и нарушению гемолиза.
Вне зависимости от вида, клиническое протекание талассемий схожи, различия зависят от степени тяжести патологического процесса.
Что такое талассемия и почему она возникает
Талассемия – это генетическое заболевание крови. Образуется недостаточное количество гемоглобина, белка, отвечающего за перенос кислорода и углекислого газа. Он состоит из четырех пептидов. 90% молекул гемоглобина содержат два альфа-глобина и два бета-глобина (гемоглобин А или A1), 2.5% содержат вместо бета-цепей дельта-цепи (гемоглобин А2), а остальные представляют собой гемоглобин A, состарившийся в процессе эксплуатации в эритроцитах (гемоглобин A3). При мутации в генах, отвечающих за синтез одного из глобинов, состав гемоглобина нарушается. Эти изменения влекут гибель эритроцитов.
Рассматриваемое заболевание впервые было описано американскими педиатрами в 1925 г. Они наблюдали 5 детей из семей итальянцев-эмигрантов. У детей обнаружили признаки гипохромной анемии тяжелого течения, значительное увеличение печени и селезенки, костные изменения. После опубликования сообщения появилась работа итальянских авторов, описавших сходную, но значительно более легкую форму течения талассемии, при которой лица, страдающие указанной патологией, доживали до зрелого возраста.
Термин «талассемия» был предложен в 1936 г. Болезнь Кули стали называть большой талассемией (thalassemia major), рассматривая ее как гомозиготную форму наследственной патологии. Впервые мысль о том, что талассемия является результатом нарушения синтеза цепей глобина, высказали независимо несколько ученых. Талассемия, при которой нарушается синтез бета-цепи глобина, называется бета-талассемией; при а-талассемии нарушается синтез а-цепи. Описаны также случаи у-, S-, 8-талассемии с нарушением синтеза соответствующих цепей глобина. Чаще встречается бета-талассемия.
Патогенез (что происходит?) во время Талассемии: При талассемии одна из цепей глобина синтезируется в малом количестве или совсем не синтезируется. В норме синтез цепей глобина сбалансирован. Количество а– и не а-цепей одинаково, и свободных цепей глобина в норме не бывает. Нарушенный синтез одной из них приводит к нарушению баланса. Цепь, которая производится в избыточном количестве, агрегирует и откладывается в эритрокариоцитах. С этим связана большая часть клинических проявлений талассемии. При а-талассемии в большинстве случаев происходит потеря участка хромосомы структурных генов, отвечающих за синтез а-цепи, она кодируется не одной, а у большинства людей двумя парами генов, расположенных в хромосоме 11. Отсутствие а-цепи у плода приводит к развитию водянки и внутриутробной смерти. Делеция в одном из 4 генов, кодирующих а-цепь, вызывает легкий дефицит а-цепи; делеция в 2 генах — более выраженный дефицит, но клинические проявления болезни во многом зависят от того, какие 2 гена не функционируют — в одной хромосоме или в разных. Неодинаково значение 2 пар генов, ответственных за синтез а-цепи: одна пара главная, другая второстепенная. Клиническая картина зависит и от того, в каких 2 из 4 генов произошла мутация. Если отсутствуют 3 гена, то у больных имеется гемоглобинопатия Н. Гемоглобин Н состоит из 4 бета-цепей. Он очень нестоек, агрегирует, и при этом развивается выраженная гемолитическая анемия. Значительно более сложен механизм бета-талассемии. Синтез бета-цепи нарушается при ряде различных заболеваний: бета-талассемии, наследственном персистировании фетального гемоглобина, гемоглобинопатии Lepore. Однако при этих заболеваниях нарушения неодинаковы. Ген, кодирующий синтез бета-цепи, располагается в хромосоме 16. В этой же хромосоме рядом с ним располагаются гены, ответственные за синтез у– и 8-цепей. При бета-талассемии, когда бета-цепь вообще не производится, не удалось обнаружить делеции гена. При талассемии имеются признаки нестабильности РНК. Основной причиной части случаев бета-талассемии является нарушение сплайсинга (процесс «сшивки» кодирующих участков). Под сплайсингом понимают изменения, которым подвергается мРНК по пути от ядра, где она синтезируется, в цитоплазму. Первичные транскрипты мРНК устроены так, что участки, кодирующие белок, чередуются с участками, его не кодирующими. Эти некодирующие участки в норме вырезаются из молекулы РНК, а кодирующие части молекулы соединяются друг с другом. Нарушение сплайсинга может приводить к нарушению стабильности мРНК. Делеция гена, ответственного за синтез 8-цепи, выявлена и при других формах патологии. Это бета-, 8-талассемия. Делеция гена 0– и 8-цепей обнаружена при наследственном персистировании фетального гемоглобина. Это своеобразное заболевание с очень легкой анемией или без нее, когда содержание фетального гемоглобина сильно увеличено (до 95-98%). В отличие от гомозиготной бета-талассемии, при которой иногда обнаруживается такое же высокое содержание фетального гемоглобина, при наследственном персистировании фетального гемоглобина нет нарушения баланса в синтезе цепей. Гемоглобинопатия Lepore — следствие делеции в генах, ответственных за синтез 8- и у-цепей.
Симптомы Талассемии: В основе клинических проявлений талассемии основное значение придается избыточному количеству цепи глобина. Так, при бета-талассемии в связи с нарушением синтеза бета-цепи оказывается много свободных а-цепей. Избыточный синтез а-цепи является главной причиной неэффективного кроветворения при бета-талассемии. Эритрокариоциты погибают в костном мозге. Гибель эритрокариоцитов и в меньшей степени ретикулоцитов и эритроцитов периферической крови в селезенке приводит к выраженному малокровию. При этом в селезенке и в печени могут формироваться очаги красного кроветворения. Интенсивное кроветворение в костях может привести к их искажению, выраженная гипоксия — к нарушению развития ребенка. Существует корреляция между глубиной анемии и избытком а-цепи при бета-талассемии. При более высоком содержании фетального гемоглобина клетки разрушаются меньше. Это связано с тем, что избыточные а-цепи находятся не в свободном, а в связанном с у-цепями состоянии в виде гемоглобина F. У некоторых больных гомозиготной талассемией нет тяжелых признаков болезни. Анемия у них не столь глубока, они могут жить без постоянных гемотрансфузий, и клинически болезнь определяют не как большая талассемия, а как промежуточная талассемия. Имеются больные более тяжелой гетерозиготной бета-талассемией. У этих больных нередко обнаруживаются включения выпавших в осадок избыточных а-цепей. Тяжесть гетерозиготной талассемии у этих больных обусловлена недостаточной способностью клеток освобождаться от избыточных а-цепей. Обычно при гетерозиготной талассемии отмечается компенсаторное повышение утилизации избыточных а-цепей. При а-талассемии в случае отсутствия синтеза а-цепи и в связи с этим гемоглобинов А, А2 и F развивается водянка плода, которая приводит его к гибели. Избыток бета-цепей при талассемии способен образовывать гемоглобин, состоящий из 4 бета-цепей, — гемоглобин Н. Клетки, содержащие гемоглобин Н, очень легко удаляются из циркуляции селезенкой. Анемия при гемоглобинопатии Н обусловлена как гемолизом периферических эритроцитов, так и нарушением синтеза глобина. Гомозиготная бета-талассемия При гомозиготной бета-талассемии, которая описана в 1925 г., клинические проявления отмечаются к концу первого или, реже, второго года жизни ребенка. В первые месяцы жизни обнаруживается лишь умеренная анемия, и далеко не всегда ясно, унаследовал ребенок болезнь от одного или обоих родителей. Гомозиготная бета-талассемия сопровождается значительным увеличением селезенки, желтушностью и сероватым оттенком кожи и слизистых оболочек, выраженной бледностью. Резкая гиперплазия кроветворного костного мозга приводит к самым разнообразным деформациям скелета: отмечаются практически квадратный череп, сильно сплющенная переносица, сужение глазных щелей. При рентгенологическом исследовании выявляют увеличение толщины губчатого слоя костей свода черепа, а также поперечную исчерченность на наружной пластинке лобной и теменной костей. Дети отстают в физическом развитии, у них снижена сопротивляемость различным инфекциям, задерживается и даже нарушается половое развитие. Содержание гемоглобина снижается до 30-50 г/л. Выявляется развитая гипохромия эритроцитов, цветовой показатель, как правило, 0,5 и ниже. Количество ретикулоцитов увеличено, раздражение красного ростка гораздо выраженнее, чем соответствующий этому раздражению ретикулоцитоз. Содержание ретикулоцитов повышается до 2,5-4%. Содержание железа сыворотки чаще всего повышено, но может не выходить за верхнюю границу нормы. В костном мозге содержание сидеробластов увеличено. Повышение уровня билирубина за счет непрямой фракции является характерной особенностью гомозиготной бета -талассемии. Вследствие чрезмерного отложения железа формируются цирроз печени, кардиосклероз, сахарный диабет. Также отмечается недоразвитие вторичных половых признаков. По степени тяжести различают: тяжелую гомозиготную талассемию, при которой больные умирают на первом году жизни, среднетяжелую, при которой дети доживают до 5-8 лет, и более легкую форму, позволяющую больным дожить до взрослого возраста. Диагноз гомозиготной бета-талассемии подтверждается исследованием содержания фетального гемоглобина в эритроцитах. При гомозиготной талассемии данный показатель увеличивается до 20-30. Гетерозиготная бета-талассемия Гетерозиготная бета-талассемия — результат наследования болезни только от одного из родителей, возможно, с бессимптомным течением либо с нерезкими клиническими проявлениями. Наиболее редко встречаются бурные клинические проявления. Жалобы больных гетерозиготной талассемией носят общий характер и проявляются слабостью, повышенной утомляемостью, понижением работоспособности, наиболее тяжело больной воспринимает физический труд. Кожные покровы у таких больных бледные, характерна небольшая иктеричность кожи и склер. Увеличение селезенки — достаточно часто встречаемый симптом гетерозиготной бета-талассемии. Показатели крови при гетерозиготной талассемии крайне разнообразны. Содержание гемоглобина может быть близко к норме. Так называемая thalassemia minima сопровождается показателями гемоглобина 110-120 г/л, либо он снижен до 90-100 г/л (thalassemia minor), редко показатель достигает 70 г/л. Анемия носит гипохромный характер. Снижение цветового показателя может быть значительным (0,5-0,7). Кроме гипохромии эритроцитов, часто выявляют анизоцитоз и пойкилоцитоз, мишеневидность эритроцитов. Также мишеневидность эритроцитов обнаруживается при железодефицитной анемии, свинцовой интоксикации, у лиц, перенесших спленэктомию. Типичным признаком талассемии служат базофильно пунктированные эритроциты. Содержание ретикулоцитов при гетерозиготной талассемии обычно повышается до 2-5%. Обнаруживается значительное раздражение красного ростка костного мозга. Нередко красный росток в 1-3 раза превышает белый. Уменьшено количество зрелых эритрокариоцитов, содержащих гемоглобин. Содержание гранул железа в костном мозге увеличено или, реже, нормально. Содержание железа сыворотки у больных гетерозиготной талассемией нормальное, реже повышенное. Запасы железа, определенные по десфераловому тесту, оказываются повышенными. В 75% случаев гетерозиготной бета-талассемией выявляется повышение уровня непрямого билирубина. Диагноз гетерозиготной бета-талассемии ставится на основании повышения содержания фракции гемоглобина А2. Содержание гемоглобина А2 при гетерозиготной бета-талассемии увеличивается до 4,2-8,9% общего количества гемоглобина. Приблизительно у половины больных обнаруживается увеличение содержания фетального гемоглобина до 2,5-7%. Важным диагностическим признаком является подобное заболевание у членов семьи больного. Алъфа-талассемия Синтез а-цепи глобина в отличие от синтеза бета-цепи контролируется двумя парами неодинаковых по значению генов, в связи с этим существуют различные варианты течения а-талассемии. При полном отсутствии а-цепи, гомозиготном нарушении функции всех 4 генов у плода не синтезируется фетальный гемоглобин, развивается водянка, в результате чего наступает смерть. Нарушение функции 1 или 2 генов приводит к анемии, как правило, нетяжелого течения. Клинические проявления а-талассемии с поражением 2 генов зависят от того, какие гены поражены, и почти полностью повторяют гетерозиготную бета-талассемию. Нередко обнаруживается увеличение селезенки, реже – печени. Определяются умеренная гипохромная анемия с мишеневидными эритроцитами и эритроцитами с базофильной пунктацией, незначительное повышение уровня ретикулоцитов, небольшая гипербилирубинемия и повышение железа сыворотки, повышение осмотической резистентности эритроцитов. Отмечается резкое раздражение красного ростка костного мозга. Как правило, кровные родственники больного имеют такую же анемию. Однако в отличие от бета-талассемии при а-талассемии не увеличивается количество фетального гемоглобина и гемоглобина А2. а-талассемию удается диагностировать только в случае изучения биосинтеза цепей глобина in vitro (в пробирке). Альфа-талассемию иногда выявляют у новорожденных: при исследовании крови обнаруживается гемоглобин Bart, лишенный а-цепи. Этот гемоглобин состоит из 4 у-цепей. Гемоглобинопатия Н является одним из вариантов а-талассемии. Она сравнительно нетяжелая, проявляется увеличением размеров как селезенки, так и печени, незначительной желтухой, в основном за счет увеличения фракции непрямого билирубина, анемией различной степени выраженности, обычно содержание гемоглобина не опускается ниже 70–80 г/л. Отмечаются выраженная гипохромия эритроцитов, их мишеневидность, базофильная пунктация. Так же как и при других формах талассемии, при гемоглобинопатии Н обнаруживаются признаки неэффективного кроветворения: резкое раздражение красного ростка костного мозга при небольшом повышении уровня ретикулоцитов. Гемоглобинопатия Н отличается от других форм талассемии множественными мелкими включениями во всех эритроцитах. Гемоглобинопатия Н впервые была описана в 1955 г. Rigas с соавторами. Данные ученые наблюдали 2 больных с гемолитической анемией, у которых при электрофорезе гемоглобина в щелочном буфере была обнаружена дополнительная фракция, движущаяся впереди гемоглобина А. Гемоглобинопатия Lepore — это формы талассемии, при которых синтез а-цепи глобина нормальный, а синтез второй цепи нарушен. Вместо нормальной бета-цепи синтезируется своеобразная цепь, состоящая из фрагментов 0– и 8-цепей. Клинические признаки гемоглобинопатии Lepore крайне похожи на таковые при гомозиготной бета-талассемии. Выявляется около 70-80% фетального гемоглобина и около 20% гемоглобина Lepore. Гетерозиготную форму гемоглобинопатии Lepore по клиническим и гематологическим признакам невозможно отличить от гетерозиготной талассемии. Содержание гемоглобина колеблется в пределах от 90 до 100 г/л, нередко увеличена селезенка. У гетерозигот содержание гемоглобина Lepore составляет 7-8%, при этом снижается содержание гемоглобина А2 и повышается содержание фетального гемоглобина. Гемоглобинопатии Lepore встречаются редко.
Лечение Талассемии: Лечение гомозиготной талассемии. Так как проявления болезни определяются гипоксией и активным кроветворением в костях, где обычно оно отсутствует, проводят переливания эритроцитов больному с раннего возраста.
Вначале применяется ударный курс лечения (за 2-3 недели 8-10 переливаний). При этом содержание гемоглобина обычно повышается до 120-140 г/л. Затем переливания делают реже, каждые 3–4 недели, из расчета 20 мл/кг. Уровень гемоглобина стараются поддерживать на 90-100 г/л. Массивные переливания при талассемии не только улучшают общее состояние, но и уменьшают изменения скелета, увеличение селезенки, улучшают развитие детей, снижают частоту тяжелых инфекций.
Частыми осложнениями данной терапии являются пирогенные реакции, в основном при применении цельной крови или недостаточно отмытых эритроцитов.
Одним из серьезных осложнений переливания является избыточное отложение железосодержащего пигмента, что приводит к существенному увеличению печени, сахарному диабету, сердечной недостаточности. Лечение гомозиготной талассемии включает обязательное использование десферала для выведения избыточного количества железа. Доза десферала (вводится внутримышечно) зависит от количества перелитых эритроцитов и возраста больного: для маленьких детей — 10 мг/кг, для подростков — 500 мг/сут. Допустимы и большие дозы — 1 г/сут. Рекомендуется сочетать десферал с 200-500 мг аскорбиновой кислоты внутрь.
Значительное увеличение селезенки, присоединение лейкопении, тромбоцитопении к признакам гипохромной анемии заставляют думать о спленэктомии.
Лечение гомозиготной талассемии указанными методами улучшает общее состояние больных, увеличивает продолжительность их жизни.
В настоящее время широко применяется пересадка костного мозга больным талассемией, в результате чего в большинстве случаев достигается стойкое улучшение.
Лечения гетерозиготной бета-талассемии в большинстве случаев не требуется. Больные чувствуют себя удовлетворительно. Содержание гемоглобина колеблется в пределах от 90 до 110 г/л. При гетерозиготной бета-талассемии переливаний крови не требуется. Удаление селезенки производится крайне редко и лишь при очень большой селезенке.
Десферал следует применять при гетерозиготной бета-талассемии, когда наблюдается высокое количество сывороточного железа и железа в моче после введения 500 мг десферала. Однако десферал лишь предотвращает сидероз (заболевание человека, вызываемое осаждением в легких пыли, содержащей железо), не приводя к повышению уровня гемоглобина.
При снижении уровня гемоглобина в связи с инфекционными заболеваниями, при беременности необходимо использовать фолиевую кислоту по 0,005 г 1-2 раза в день, так как при неэффективном кроветворении, связанном с талассемией, потребляется значительное количество фолиевой кислоты.
При гетерозиготной талассемии противопоказаны препараты железа, так как всегда есть некоторый избыток железа без клинических проявлений гемосидероза. Однако больные, принимающие препараты железа, чувствуют себя значительно хуже. Наиболее опасно при талассемии парентеральное введение препаратов железа: больные, находившиеся в удовлетворительном состоянии, могут умереть от тяжелого гемосидероза, в частности от недостаточности кровообращения, связанной с сидерозом миокарда.
Лечение а-талассемш практически не отличается от лечения гетерозиготной талассемии, за исключением гемоглобинопатии Н.
При гемоглобинопатии Н в связи с нестабильностью этого гемоглобина и выпадением его в осадок имеются признаки не только неэффективного кроветворения, но и выраженного разрушения эритроцитов периферической крови. Это разрушение идет преимущественно в селезенке, в результате чего она увеличивается. Основным методом лечения гемоглобинопатии Н при выраженной анемии является удаление селезенки.
Иллюстрации с сайта: © 2011 Thinkstock
Причины и факторы риска развития заболевания
Талассемия – заболевание наследственное, то есть мутация передается от родителя ребенку. Если кто-то из родственников страдал от этой патологии, риск развития заболевания повышается.
Второй фактор риска – этническая принадлежность. Больше всего талассемия распространена в Африке, Средней Азии и странах Средиземноморья, где она и была открыта (в переводе с греческого “талассемия” — это “морская анемия”). Различают два основных типа талассемии: альфа и бета. В первом случае мутация затрагивает гены, отвечающие за синтез альфа-глобинов, во втором – бета.
Альфа-глобины кодируются четырьмя генами. Тяжесть заболевания будет зависеть от количества патологически измененных участков ДНК:
- 1 измененный ген – бессимптомная форма. Но при ней человек становится носителем заболевания и может передать его своим детям;
- 2 гена – легкое течение болезни;
- 3 гена – тяжелое течение болезни;
- 4 гена – редкий тип заболевания, который плохо совместим с жизнью. Большинство плодов гибнет еще в период внутриутробного развития, а родившиеся малыши, в основном, умирают вскоре после рождения или нуждаются в пожизненной терапии. В отдельных случаях удается их вылечить путем пересадки костного мозга.
Бета-глобины кодируются одним геном, который локализуется в 11-й хромосоме. Если дефектный ген содержится только в одной хромосоме из пары, заболевание протекает легко (малая талассемия). Повреждение обеих хромосом приводит к очень серьезному заболеванию, известному как большая талассемия или болезнь Кули (анемия Кули).
Немного истории
Болезнь была впервые описана в 1925 году американскими врачами-педиатрами, которые при лечении эмигрантов из Италии выявили одинаковую клиническую картину у детей с тяжелым малокровием, увеличением печени и селезенки, изменениями костей.
Затем появились работы, описывающие более легкое течение болезни у взрослых пациентов. Термин «талассемия» предложили в 1936 году. Дословно он означает «болезнь морского побережья». Высказана мысль о связи патогенеза заболевания с нарушением синтеза глобиновых цепочек.
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников — бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Диагностика
Диагностика талассемии должна начинаться с генетической консультации супругов перед зачатием ребенка. Во время беременности при необходимости проводят анализ амниотической жидкости. Уже на ранних сроках в эритроцитах плода можно обнаружить характерные изменения.
Порой внешний осмотр и выяснение семейного анамнеза позволяют заподозрить болезнь у ребенка без лабораторной диагностики.
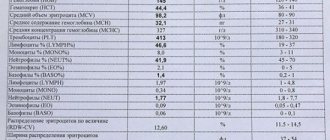
В анализе крови выявляют:
- снижение уровня гемоглобина до 30–50 г/л при гомозиготном варианте и до 90 –110 г/л при гетерозиготном;
- низкий цветовой показатель (менее 0,5), который образуется малым насыщением клеток гемоглобином);
- рост ретикулоцитов (предшественников эритроцитов) до 4%.
При просмотре окрашенного мазка под микроскопом обращают внимание на следующее:
- наличие слабо окрашенных (гипохромных) эритроцитов;
- изменение размеров и формы красных клеток, эритроциты приобретают формы овала, серпа, шара.
Одна из характерных разновидностей серповидных клеток — дрепаноциты.
Дрепаноциты образуются при любых гемоглобинопатиях, содержат особую разновидность (S гемоглобин), который может в условиях низкой концентрации образовывать полимеры и изменять форму оболочки клетки
Биохимические тесты показывают нарушенный обмен железа, как при гемолитической анемии:
- высокий уровень свободного билирубина;
- повышенную концентрацию железа в сыворотке;
- сниженную способность к связыванию железа.
Исследование особенностей гемоглобина проводят с помощью ацетат-целлюлозной пленки. Подобный анализ позволяет определить количественный уровень фракций. Гомозиготная бета-талассемия отличается увеличенным уровнем фетального гемоглобина (в норме он содержится у плода, а у взрослого человека всего до 1%), неспособного переносить кислород.
Использование метода полимеразной цепной реакции позволяет изучить строение полипептидных цепочек гемоглобина.
Генетический анализ выявляет мутацию в одиннадцатой паре хромосом при β-талассемии, другие специфические изменения, типичные для прочих форм.
Исследование пунктата костного мозга проводится для выявления повышенного содержания незрелых эритроцитов в виде сидеробластов.
Рентгенологическое исследование способствует обнаружению дефектов костной ткани:
- участков остеопороза (сниженной плотности);
- увеличенной массы костей черепа;
- поперечной исчерченности на мелких костях кистей и стоп;
- на рентгенограмме черепа при большой форме β-талассемии видно типичное игольчатое поражение надкостницы, которое называется симптомом «волосатого черепа».
Ультразвуковое исследование подтверждает увеличение печени и селезенки, помогает обнаружить камни в желчевыводящей системе.
Лабораторные данные более отчетливо выражены при β-талассемии. Другие формы болезни не дают четкой картины.
Заболевания, с которыми необходимо проводить дифференциальную диагностику:
- железодефицитная анемия,
- серповидно-клеточная анемия,
- гемолитическая аутоиммунная анемия,
- наследственный микросфероцитоз.
Лечение талассемии
Определяется типом и степенью тяжести. При умеренно выраженных симптомах лечение не назначают. Время от времени проводят только переливание крови. В основном это нужно после операций, родов или для предотвращения возможных осложнений. Люди с бета-талассемией требуют более частых переливаний крови. Для нормализации избыточного уровня железа им также назначают специфические препараты, которые связывают и выводят железо.
При выраженной и тяжелой формах болезни существует два способа лечения:
- частые переливания крови (раз в несколько недель), которые сочетают с приемом препаратов, выводящих лишнее железо из организма;
- пересадка костного мозга – единственный метод, который помогает полностью излечить человека от талассемии. К сожалению, далеко не всегда трансплантация бывает успешной.
Лекарственные препараты при талассемии назначают только для коррекции симптомов и осложнений. Медикаментозной терапии самого заболевания не существует.
Разновидности заболевания
Все виды заболевания связаны с кислородной недостаточностью, наступившей вследствие разрушения эритроцитов — единственных клеток, обеспечивающих газообмен в органах и тканях.
В зависимости от сбоя синтеза полипептидной цепочки различают:
- альфа-талассемию;
- бета-талассемию.
Наиболее распространена бета-талассемия. Она характеризуется избыточным накоплением α-цепочек глобина.
Выявлены также редкие формы болезни, которые названы гамма- и дельта-талассемией. В группу включены:
- некоторые гемоглобинопатии;
- гомозиготная альфа-талассемия с водянкой плода;
- смешанный вид β- и δ-талассемии, когда поражаются одновременно дельта- и бета-цепи.
Каждая форма имеет свои типичные клинические проявления и отличия. Но в зависимости от тяжести течения принято выделять:
- хроническую легкую — люди доживают до старости;
- хроническую среднетяжелую — пациенты не переживают детский возраст;
- тяжелую — ребенок погибает в период новорожденности.
Осложнения талассемии
Возможные осложнения заболевания:
- излишек железа, которое входит в состав гемоглобина. Накопление этого элемента в организме приводит к поражению сердца, печени, эндокринной системы;
- подверженность инфекциям. Особенно актуально для пациентов, которым удалили селезенку;
- деформация костей, связанная с увеличением объема костного мозга. Чаще всего этот процесс затрагивает кости черепа, реже конечностей. Они истончаются и чаще ломаются;
- спленомегалия — увеличение селезенки, где в основном и гибнут дефектные эритроциты. Если селезенка увеличена очень сильно, ее удаляют. Эта операция называется спленэктомия;
- задержка роста и полового созревания;
- сердечные болезни (хроническая сердечная недостаточность и аритмии) могут развиться при тяжелом течении заболевания.
Терапия и профилактика
Для лечения тяжелых форм талассемии используют переливание крови – для повышения количества эритроцитов и восстановления кислородного транспорта, а также хелатирование – для выведения свободной формы железа, которая не встроилась в «неправильный» гемоглобин. При тяжелой форме бета-талассемии переливание крови необходимо часто – каждые 3-4 недели. Однако сейчас такая терапия позволяет пациентам вести нормальный образ жизни и восстанавливает продолжительность жизни. Для легких форм талассемии показания проще – врачи советуют соблюдать здоровый образ жизни и больше двигаться. Занятия спортом позволяют поддерживать минерализацию костей скелета и препятствуют их деформации.
Как сейчас лечат бета-талассемию?
Бета-талассемию по идее можно вылечить процедурой трансплантации гемопоэтических стволовых клеток, однако, увы, подходящих доноров критически мало: таковых можно найти где-то для 25–30% пациентов. Опять же, ввиду высокого риска отторжения трансплантата и реакции «трансплантат против хозяина», эту процедуру обычно проводят только маленьким детям.
И потому в большинстве случаев заболевание предполагает хроническую терапию, которая включает регулярные гемотрансфузии эритроцитарной массы, хелирующие железо агенты, спленэктомию, индуцирование фетального гемоглобина. Качество жизни пациентов остается весьма низким.
На заметку
- Талассемия – тяжелое наследственное заболевание, возникающее из-за мутаций в генах гемоглобина. Для нее свойственны слабость и головокружения, болезни печени и сердца, увеличение размеров селезенки, а также деформация костей.
- Выделяют два вида болезни – альфа- и бета-талассемию. В среднем в мире носителей мутаций, характерных для обоих видов, – около пяти процентов, в России же – всего около одного процента.
- Диагностируют талассемию при помощи анализов крови, УЗИ и генетического теста.
- Для лечения тяжелых форм используют частые переливания крови и выведение свободных форм железа. Для легких форм оказывается достаточно поддержания здорового образа жизни.
Источники:
- Практическая медицина, Гемоглобинопатии и талассемические сидромы, 2015
- Subcellular Biochemistry, Hemoglobin: Structure, Function and Allostery, 2020
- ФГУ ФНКЦ ДГОИ Росздрава, Эпидемиология гемоглобинопатий в Москве, 2008
- American Family Physician, Alpha and Beta Thalassemia, 2009
- NCBI, Thalassemia, 2021
Теги
- Генетика
- Здоровье